Methods
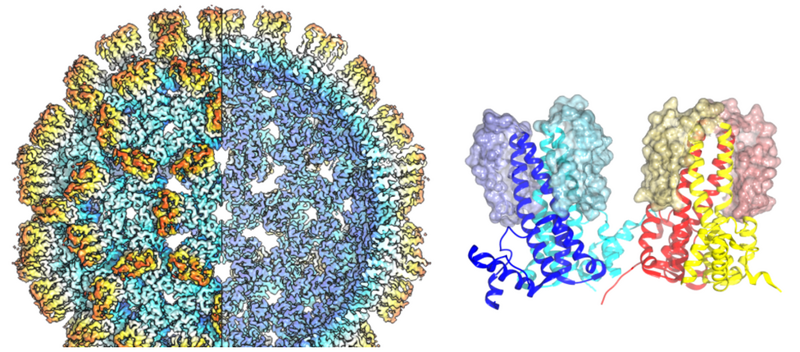
Understanding how cells function requires a comprehensive understanding of both their proteomic composition and their structural organisation. Two central technologies of modern quantitative biology have been established at the Chair of Biochemistry II and form the basis for new biological findings. High-resolution mass spectrometry in Prof Bettina Warscheid 's working group makes it possible to identify and quantify thousands of proteins from tissue samples through to isolated cell groups, thereby revealing functional and structural networks in cells. In addition, cryo-electron microscopy, established in Prof Bettina Böttcher 's working group, allows the three-dimensional structure of protein complexes to be determined, thus laying the foundation for understanding how they function and for developing targeted interactors (e.g. in the form of drugs).
Mass spectrometry and functional proteomics
Prof Warscheid's working group has various mass spectrometers (for details see bioMS technology platform) on which experiments can be carried out using almost all currently available MS methods. The methodological focus of the working group is divided into
- Quantitative proteomics (both steady state and time-resolved)
- Identification and localisation of post-translational modifications
- Structural proteomics
These methodological focuses are complemented by bioinformatic expertise in the form of our own data analysis and processing pipelines, which are mainly written in Python.
 on a system-wide scale. Key to our in-depth analysis of protein interaction and signalling networks, membrane protein assemblies and dynamic organellar proteomes is the use of quantitative high-resolution MS technologies in combination with computational proteomics approaches and statistical data analysis.")
Methods of quantitative proteomics include label-free quantification via data-dependent and data-independent analysis, chemical labelling e.g. dimethyl tags or TMT as well as metabolic labelling (SILAC). All these methods allow a different weighting of quantitative accuracy, costs and complexity of data analysis. Different properties of the available mass spectrometers are used, e.g. real-time search with synchronous precursor selection for MS (RTS SPS MS3) for TMT quantification or narrow window DIA on the ASTRAL analyser for precise label-free quantification.
For the identification and localisation of phosphorylation and ubiquitylation, we apply similar principles as for label-free analysis, but particularly labile modifications sometimes require very gentle fragmentation conditions. For this purpose we use e.g. electron transfer dissociation (ETD), an orthogonal method to classical higher energy collisional dissociation (HCD). For the precise quantification of known modifications under different experimental conditions, we use both offline fractionation and targeted MS, i.e. the definition of MS1/MS2 transitions that are permanently monitored by the device.
Structural proteomics comprises methods that define the local relationship of proteins or protein complexes to each other. This ranges from global approaches such as complexome profiling in which the subcellular localisation of proteins is determined on the basis of correlations, e.g. during density gradient centrifugation, to proximity proteomics, i.e. methods such as bioID or APEX in which spatial proximity to a modified protein is encoded in the form of a chemical label, through to chemical cross-linking in which proteins that are in close proximity are permanently covalently cross-linked so that not only the interaction but also the area of interaction can be defined. For isolated protein complexes, we also use native MS of intact protein complexes, which allows us to achieve resolutions corresponding to mass differences in the Dalton range using an Orbitrap analyser.
The central facility for cryo-electron microscopy provides easy access to a Titan-Krios G3 with an X-FEG source, 300 kV, Selectris energy filter and a Falcon IVi camera with direct electron detection. The setup is optimised for high-resolution image acquisition with high throughput for structure determination using single-particle image analysis. Image acquisition can be performed in linear or counting mode. Acquisition is automated with EPU and can run for several days without further intervention. Typically, data acquisition takes 1-3 days. Longer sessions can be performed if required.


History
The facility was commissioned in 2018. In January 2023, support for single particle image processing (3 workstations) was added, providing a computing environment for small processing projects (max. 6 weeks), training and processing on behalf of users. In September 2023, the camera system was upgraded to a Falcon IVi with Selectris energy filter for faster high-resolution acquisition in counting mode.