Methoden
Die Erschließung der Funktionsweise von Zellen erfordert ein umfassendes Verständnis sowohl ihrer proteomischen Zusammensetzung als auch ihrer strukturellen Organisation. Am Lehrstuhl für Biochemie II sind zwei zentrale Technologien der modernen, quantitativen Biologie etabliert und bilden die Basis für neue biologische Erkenntnisse. Hochauflösende Massenspektrometrie in der Arbeitsgruppe von Prof. Bettina Warscheid erlaubt es tausende von Proteinen aus Gewebeproben bis hin zu isolierten Zellgruppen zu identifizieren, quantifizieren und damit funktionelle und strukturelle Netzwerke in Zellen aufzudecken. Durch Kryo-Elektronenmikroskopie, etabliert in der Arbeitsgruppe von Prof. Bettina Böttcher, kann schließlich die dreidimensionale Struktur von Proteinkomplexen bestimmt und damit die Grundlage für das Verständnis von deren Funktionsweise und die Entwicklung von gezielten Interaktoren (z.B. in Form von Arzneimitteln) gelegt werden.
Massenspektrometrie und funktionelle Proteomik
In der Arbeitsgruppe von Prof. Warscheid stehen verschiedene Massenspektrometer (Details siehe bioMS Technologieplattform) auf denen Experimente mit fast allen aktuell verfügbaren MS-Methoden absolviert werden können. Die methodischen Schwerpunkte der Arbeitsgruppe gliedern sich in
- Quantitiative Proteomik (sowohl steady state als auch zeitaufgelöst)
- Identifzierung und Lokalisierung von post-translationalen Modifikationen
- Strukturelle Proteomik
Diese methodischen Schwerpunkte werden komplementiert durch bioinformatische Expertisen in Form eigener Datenanalyse- und Prozessierungspipelines, die überwiegend in Python geschrieben sind.
 auf einem System zu untersuchen. Der Schlüssel zu unserer gründlichen Analyse von Proteininteraktions- und Signalnetzwerken, Membranproteinanordnungen und dynamischen organellaren Proteomen ist die Verwendung quantitativer hochauflösender MS-Technologien in Kombination mit computergestützten Proteomik-Ansätzen und statistischer Datenanalyse.")
Methoden der quantitativen Proteomik umfassen labelfreie Quantifizierung über datenabhängige und -unabhängige Analyse, chemisches Labeling z.B. Dimethyltags oder TMT sowie metabolisches Labeling (SILAC). All diese Methoden erlauben eine unterschiedliche Gewichtung von quantitativer Genauigkeit, Kosten und Komplexität der Datenanalyse. Dabei kommen unterschiedliche Eigenschaften der vorhandenen Massenspektrometer zum Einsatz, z.B. real-time search mit synchronous precursor selection für MS (RTS SPS MS3) für die TMT-Quantifizierung oder narrow window DIA am ASTRAL Analysator für die präzise labelfreie Quantifizierung.
Bei der Identifizierung und Lokalisierung von Phosphorylierungen und Ubiquiitylierung wenden wir ähnliche Prinzipien an, wie bei der labelfreien Analyse, allerdings benötigen besonders labile Modifikationen z.T. sehr sanfte Fragmentierungsbedingungen. Hierzu nutzen wir z.B. electron transfer dissociation (ETD), eine orthogonale Methode zur klassischen higher energy collisional dissociation (HCD). Zur präzisen Quantifizierung von bekannten Modifikationen bei unterschiedlichen Versuchsbedingeungen nutzen wir sowohl offline Fraktionierung als auch targeted MS, also die Definition von MS1/MS2 Übergängen die permanent vom Gerät überwacht werden.
Die strukturelle Proteomik umfasst Methoden, die die örtliche Beziehung von Proteinen oder Proteinkomplexen zueinander definierten. Dies reicht von globalen Ansätzen wie complexome profiling, bei dem die subzelliuläre Lokalisation von Proteinen anhand von Korrelationen z.B. bei der DIchtegradientenzentrifugation ermittelt wird, über proximity proteomics, also Methoden wie bioID oder APEX, bei denen eine räumliche Nähe zu einem modifizierten Protein in Form einer chemischen Markierung codiert wird bis hin zu chemischem cross-linking, bei dem Proteine, die sich in räumlicher Nähe befinden, permanent kovalent vernetzt werden, sodass nicht nur die Interaktion, sondern die Interaktionsfläche definiert werden kann. Für isolierte Proteinkomplexe verwenden wir außerdem native MS von intakten Proteinkomplexen, womit wir unter Verwendung eines Orbitrap-Analysators Auflösungen erreichen, die Massenunterschieden im Dalton-Bereich entsprechen.
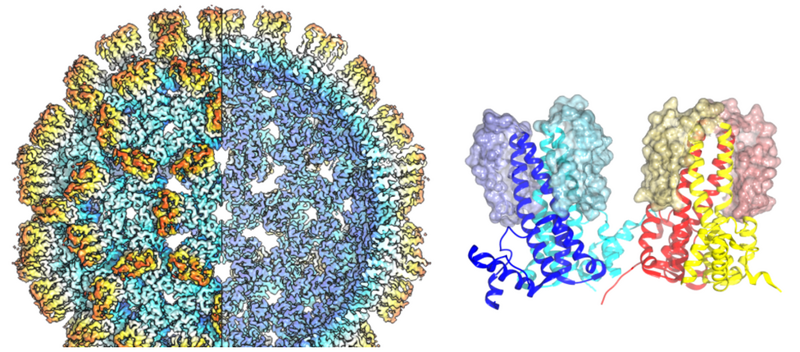
Kryo-Elektronenmikroskopie
Die zentrale Einrichtung für Kryo-Elektronenmikroskopie bietet einen erleichterten Zugang zu einem Titan-Krios G3 mit einer X-FEG-Quelle, 300 kV, Selectris-Energiefilter und einer Falcon IVi-Kamera mit direkter Elektronendetektion. Der Aufbau ist für hochauflösende Bilderfassung mit hohem Durchsatz zur Strukturbestimmung durch Einzelpartikel-Bildanalyse optimiert. Die Bildaufnahme kann im linearen Modus oder im Zählmodus erfolgen. Die Aufnahme wird mit EPU automatisiert und kann über mehrere Tage ohne weitere Eingriffe laufen. Typischerweise dauert die Datenerfassung 1-3 Tage. Bei Bedarf können auch längere Sitzungen durchgeführt werden.


Geschichte
Die Anlage wurde 2018 in Betrieb genommen. Im Januar 2023 wurde die Unterstützung für die Verarbeitung von Einzelpartikelbildern (3 Workstations) hinzugefügt, die eine Rechenumgebung für kleine Verarbeitungsprojekte (max. 6 Wochen), Schulungen und Verarbeitung im Auftrag von Nutzern bietet. Im September 2023 wurde das Kamerasystem auf eine Falcon IVi mit Selectris-Energiefilter für eine schnellere hochauflösende Erfassung im Zählmodus aufgerüstet.